Синдром фанкони у взрослых клиника лечение патогенез. Синдром фанкони у детей говорит о мутации генов. Эффективные методы лечения
Синдром (болезнь) де Тони - Дебре - Фанкони - одно из рахитоподобных заболеваний .
История
Данный почечный синдром был идентифицирован швейцарским педиатром Фанкони среди ранее описанных другими исследователями отдельных частей заболевания. В 1931 г. он описал у ребёнка с карликовостью и рахитом глюкозурию и альбуминурию, 2 года спустя де Тони добавил к клинической картине гипофосфатемию, а вскоре Дебре описал аминоацидурию.
Этиопатогенез
Тип наследования - аутосомно-рециссивный, выделена также аутосомно-доминантная форма с локализацией гена на хромосоме 15q15.3. Экспрессивность мутантного гена в гомозиготном состоянии значительно варьирует. Встречаются спорадические случаи, обусловленные свежей мутацией. Полагают, что в основе болезни лежат генетически обусловленные дефекты ферментативного фосфорилирования в почечных канальцах (комбинированная тубулопатия), дефицит ферментов 2-го и 3-го комплексов дыхательной цепи - сукцинатдегидрогеназного и цитохромоксидазного. Ряд авторов относит данное заболевание к разряду митохондриальных болезней .
Вышеописанные причины приводят к нарушению процессов энергообеспечения транспорта фосфатов, глюкозы и аминокислот в почечных канальцах и повышенной их экскреции с мочой, а также к расстройству механизмов поддержания кислотно-основного равновесия. Развивающийся метаболический ацидоз и недостаток фосфорных соединений способствует нарушению формирования костной ткани по типу остеомаляции и рахитоподобных изменений скелета.
Клиническая картина
Первые признаки появляются во второй половине первого года жизни, развёрнутый симптомокомплекс формируется ко второму году жизни. Иногда бывает манифестация в 6-7 лет. Начальные признаки - жажда , полиурия , рвота , иногда длительный субфебрилитет . На втором году выявляют отставание физического развития и костные деформации нижних конечностей (вальгусные или варусные), грудной клетки, предплечий и плечевых костей, снижение мышечного тонуса. Рентгенологически при этом определяют системный остеопороз различной степени выраженности, истончение коркового слоя трубчатых костей, разрыхление зон роста, отставание темпов роста костной ткани от биологического возраста ребёнка. Кости становятся ломкими.
В зависимости от тяжести клинических проявлений и метаболических расстройств выделяют два клинико-биохимических варианта болезни. Первый характеризуется значительной задержкой физического развития, тяжёлым течением заболевания с выраженными костными деформациями и нередко переломами костей, резкой гипокальциемией (1,6-1,8 ммоль\л), снижением абсорбции кальция в кишечнике. При втором варианте отмечают умеренную задержку физического развития, лёгкое течение с незначительными костными деформациями, нормокальциемию и нормальное усвоение кальция в кишечнике.
Биохимические нарушения
- снижение уровня кальция в крови;
- снижение уровня фосфора в крови;
- повышение уровня щелочной фосфатазы;
- развитие метаболического ацидоза (рН=7,35-7,25; ВЕ=-10…-12 ммоль\л) за счет дефекта реабсорбции бикарбонатов в проксимальных канальцах;
- нормальная экскреция кальция с мочой;
- повышение клиренса фосфатов мочи, всасывание фосфатов в кишечнике не страдает;
- развитие глюкозурии (20-30 г\л и выше);
- развитие генерализованной гипераминоацидурии;
- нарушение функций аммониоацидогенеза - снижение титрационной кислотности, повышение рН мочи больше 6,0;
- развитие гипокалиемии .
Дифференциальный диагноз
Болезнь де Тони-Дебре-Фанкони необходимо дифференцировать с вторичным синдромом, обнаруживаемым при других наследственных и приобретённых заболеваниях (
Синдром Фанкони представляет собой системное нарушение обмена веществ, при котором выявляется дисфункция почек. Они утрачивают способность обратного всасывания и возвращения в кровоток аминокислот, глюкозы, солей кальция и фосфора, выводящихся с мочой. Это врожденный или приобретенный органический дефект. Встречается он чрезвычайно редко, 1 случай приходится на 350 тысяч новорожденных. Однако эта дисфункция крайне тяжело отражается на состоянии организма.
Ее называют также синдромом де-Тони-Дебре-Фанкони, фосфат-диабетом, наследственным синдромом Фанкони. Одни ученые полагают, что эта патология возникает, когда в клетках резко уменьшаются запасы АТФ (аденозинтрифосфорной кислоты). Другие выдвигают версию о том, что рахитичная деформация костей происходит из-за дефицита фосфора, сбоя в кислотно-щелочном балансе либо из-за обоих этих факторов. Третьи считают, что дисфункция почек обусловлена не биохимическим, а структурным дефектом. Точная же причина до сих пор не установлена.
При такой патологии поражаются почечные проксимальные канальцы. Концентрация соединений кальция и калия в кровотоке сохраняется в пределах нормы. Реакция мочи - щелочная или нейтральная. Из-за значительных потерь гидрокарбонатов развивается ацидоз. Это опасное состояние, при котором происходит нарушение кислотно-щелочного баланса и «закисление» организма. Ацидоз, как правило, вызывает любая диффузная болезнь почек.
Врожденная патология может иметь различные степени тяжести. При наличии 3 биохимических дефектов диагностируют полную, а при 2 — неполную дисфункцию почек. Некоторые ученые считают, что наследственный синдром Фанкони - самостоятельное рахитоподобное заболевание. Однако чаще всего оно сопутствует другим врожденным патологиям. Это могут быть:
- цистиноз (заболевание, связанное с кристаллизацией аминокислоты цистин в тканях);
- болезнь Вильсона-Коновалова (патология, вызываемая отложениями меди в почках, печени, мозге);
- галактоземия (нарушение преобразования простого сахара в глюкозу);
- синдром Лоу (патология, проявляющаяся задержкой роста, психического развития, катарактой и глаукомой);
- тирозинемия (пассивность фермента печени, расщепляющего аминокислоту тирозин);
- непереносимость фруктозы.
Приобретенная дисфункция, названная именем Фанкони, может возникнуть из-за таких факторов:
- медикаментов, токсичных для почек (Аспирина, Гентамицина, просроченного Тетрациклина, антивирусных средств Цидофовир, Диданозин, противоопухолевых препаратов Стрептозоцин, Ифосфамид и др.);
- отравления солями тяжелых металлов, другими агрессивными химическими соединениями;
- острого дефицита витамина D;
- множественной миеломы (рака крови);
- амилоидоза (нарушения белкового обмена);
- трансплантации почки.

Симптомы болезни у детей
Наиболее ярко проявляется врожденный синдром Фанкони у детей. Его симптомы дают о себе знать уже на первом году жизни малышей. Это:
- полиурия (учащенное мочеиспускание с большим количеством выводящейся из организма жидкости);
- полидипсия (патологически сильная жажда);
- повышенная температура;
- судороги;
- частая рвота без видимых причин;
- затяжные запоры;
- кожная сыпь;
- отечность суставов;
- увеличение почек, лимфоузлов, селезенки;
- предрасположенность к инфекциям.

Из-за ежедневного выведения из организма с жидкостью витаминов, микроэлементов, глюкозы ребенок отстает в умственном и физическом развитии. У него искривляются кости ног, становятся дряблыми, а потом и вовсе атрофируются мышцы. Нередко дети утрачивают способность ходить самостоятельно. К 12–13 годам развивается хроническая почечная недостаточность. Иногда ухудшается зрение. Кроме того, при синдроме Фанкони симптомы патологии могут свидетельствовать о сочетанных врожденных дефектах сосудов и сердца, органов пищеварения, мочеполовой системы.
Симптомы болезни у взрослых
Чаще всего отмечаются:
- полиурия;
- гипостенурия (снижение относительной плотности мочи);
- ломота в костях;
- остеомаляция (размягчение костных тканей и утрата ими прочности);
- мышечная слабость;
- ускоренное прогрессирование гипертонии;
- хроническая почечная недостаточность (если отсутствует лечение).
Полиурия при синдроме Фанкони у взрослых не выражена резко, этим она существенно отличается от избыточного количества мочи при несахарном диабете. Чаще всего данный синдром становится проявлением множественной миеломы или болезни Вальденстрема (злокачественного поражения кроветворной системы). Кроме умеренной полиурии, нарушение деятельности почек проявляется снижением их концентрационной функции, появлением белка в моче.
О том, есть ли синдром Фанкони или нет, можно судить по таким характерным симптомам, как высокая концентрация в моче аминокислот, глюкозы и фосфатов. Повышенное выведение глюкозы с мочой при этой патологии обусловлено дисфункцией почек. Однако концентрация сахара натощак и уровень гипергликемии у этих больных, как правило, в пределах нормы.
Полиурия всегда сопровождается мышечной слабостью, которая острее всего ощущается в конечностях. Возникает она из-за дефицита калия. Кроме того, практически всегда больных донимает ломота в костях.

Симптомы ацидоза у взрослых и детей одинаковы. Это:
- потеря аппетита;
- беспричинная рвота;
- запоры или поносы;
- кислый запах, исходящий от кожи или изо рта;
- снижение давления;
- головные боли;
- одышка;
- бессонница;
- упадок сил.
Тем не менее врачи отличают взрослый ацидоз от детского. Считается, что у младенцев это всегда врожденная патология. У взрослых же она может быть как дальнейшим развитием детской болезни, так и приобретенным осложнением вследствие поражения почек. Симптомы ацидоза нередко сочетаются с признаками интерстициального нефрита и мочекаменной болезни. Конкременты в почках образуются из-за больших потерь кальция с мочой. В тяжелых случаях выявляются симптомы остеопороза и остеомаляции.
Диагностика патологии
Выявление синдрома де-Тони-Дебре-Фанкони начинается со сбора анамнеза, осмотра пациента и лабораторных анализов. Характерные признаки патологии в биохимической формуле крови - низкое содержание кальция, фосфора, калия и натрия, а также глюкозы, аланина, глицина, глютаминовой кислоты. Зато в моче - высокий уровень фосфора и глюкозы, присутствуют белок и лейкоциты.
При значительной потере гидрокарбонатов с мочой и чрезмерном накоплении кислот в организме диагностируется метаболический ацидоз. Практически у всех пациентов наблюдается превышение уровня пировиноградной и молочной кислот в крови. Выявляется и падение активности ферментов энергетического обмена.
При инструментальной диагностике обязательно проводят УЗИ почек и мочеточников. Нефробиопсия (исследование миниатюрных образцов почек) позволяет увидеть деформацию проксимальных канальцев, которые вытянуты наподобие лебединой шеи. На поздних стадиях заболевания выявляется атрофия почечных клубочков.

Рентгенография деформированных нижних конечностей помогает обнаружить перерождение костных тканей. У детей нередко встречаются скрытые переломы эпифизарных хрящей, из-за которых кости впоследствии перестают расти в длину и становятся асимметричными. В большеберцовых костях выявляются новообразования, напоминающие шпоры.
На поздних стадиях развития синдрома диагностируется остеопороз. Велик риск переломов трубчатых костей. Стадию остеопороза определяют методом рентгеноденситометрии. Низкая минерализация костных тканей выявляется при исследовании их образцов, получаемых тоже с помощью биопсии.
Главные критерии диагностики:
- Значительный дефицит веса и роста ребенка.
- Слабость статико-моторных функций.
- Рахитоподобные деформации скелета (деградация структуры костных тканей подтверждается рентгенологически).
- Электролитные нарушения.
При синдроме Фанкони у взрослых врачи обязательно проводят дифференциальную диагностику, чтобы исключить схожие с ним патологии. Это:
- наследственные заболевания (цистиноз, галактоземия, болезнь Вильсона-Коновалова, тирозинемия и др.);
- хронический пиелонефрит;
- сахарный диабет;
- вторичный гиперпаратиреоз;
- множественная миелома;
- отравления лекарственными препаратами, агрессивными химическими веществами;
- обширные ожоги.
Лечение патологии
Идеально, когда синдром Фанкони лечат генетик, гематолог, но такие специалисты есть далеко не в каждом медучреждении. Чаще всего этой патологией занимаются нефролог или уролог. Если наблюдаются признаки гиперпаратиреоза (гиперсекреции паращитовидных желез), требуется консультация эндокринолога. При ухудшении зрения необходимо обследоваться у офтальмолога.

Тактикой лечения предусматриваются:
- Восполнение дефицита электролитов.
- Устранение нарушений кислотно-щелочного баланса.
- Терапия почечной недостаточности.
- Снятие болезненной симптоматики.
При медикаментозном лечении применяются:
- Кальцитриол, Оксидевит и другие препараты витамина D;
- кальция глюконат;
- Фитин, кальция глицерофосфат, алюминия фосфат;
- ощелачивающие растворы Бицитра, Полицитра;
- Индометацин, Метилтестостерон, Гипотиазид (при тяжелом поражении проксимальных канальцев);
- Панангин, Аспаркам;
- Цистин, Меркаптамин;
- антибиотики;
- кортикостероиды и др.
Поскольку синдром Фанкони имеет хронический характер, лечение проводится длительное время, большими курсами после обязательных перерывов. Зачастую удается приблизить к норме обмен веществ, снять остроту проявления недуга и предотвратить опасные осложнения. Но полностью избавиться от синдрома де-Тони-Дебре-Фанкони чрезвычайно сложно, он часто дает рецидивы.
Терапия синдрома направлена на устранение ацидоза, восполнение запасов калия, бикарбоната кальция, фосфатов и других электролитов. Важнейшую роль при этом играет лечебная диета. Необходимо обильное питье и сокращение количества соли. Принимать пищу следует малыми порциями, но часто. Углеводы нужно строго дозировать, чтобы не было избытка глюкозы в кровотоке.
При нарушении почечной функции, отвечающей за обратное всасывание полезных веществ, обнаруживается синдром Фанкони. Второе название состояния - синдром де Тони-Дебре-Фанкони. При патологии наблюдается глюкоза, белок с , происходят метаболические нарушения. Недуг передается по наследству. Диагноз ставится по клиническим результатам. Цели лечения - возмещение дефицитных состояний, устранение почечной недостаточности.
У детей сидром де Тони-Дебре-Фанкони вызывает рахит, физическое отставание, ослабленности мышечной ткани.
Описание и причины
Недуг де Тони-Дебре-Фанкони проявляется выраженной канальцевой дисфункцией, в результате которой расстраивается процесс обратного поглощения полезных для организма веществ и ионов. Одновременно повышается выделение бикарбонатов, обнаруживаются общие метаболические сбои. Особенности:
- падение содержания кальция с фосфором в кровяной плазме;
- сбой в кислотно-основном балансе, что проявляется низкой кислотностью и бикарбонатов в крови;
- нормальное выделение кальция в виде конечного метаболита с мочой при повышенном показатели естественного очищения организма от фосфатов;
- патологически высокая активность щелочной фосфатазы;
- появление сахара в моче;
- общее нарушение выработки и выведения продуктов синтеза аминокислот;
- увеличенные объемы выводимой мочи с пониженной плотностью.
 Недуг де Тони-Дебре-Фанкони у детей.
Недуг де Тони-Дебре-Фанкони у детей.
Синдром Фанкони развивается и у детей, и у взрослых. Различают врожденный и приобретенный типы патологии. По степени тяжести симптомов, а также по типу и выраженности метаболических расстройств различают 2 формы болезни:
- Первый тип проявляется в виде выраженного отставания в физическом развитии с явной деформацией костей скелета, тяжелой клинической картиной, частыми переломами на фоне резкой недостачи кальция вследствие слабого всасывания вещества в кишечнике.
- Второй вариант проявляется в виде средней степени задержки физического развития, облегченным проявлением симптомов со слабой костной деструктцией, достаточным уровнем кальция.
Причины болезни
Провоцирующие факторы делятся на те, что вызывают приобретенную форму синдрома де Тони-Дебре-Фанкони, и те, что отвечают за появление врожденной патологии. Провокаторами вторичного нарушения признаны такие состояния:
- непереносимость фруктозы;
- нехватка клеточных ферментов;
- хроническое отравление токсинами;
- острый дефицит витамина D.
Самостоятельно синдром вызван одной плохой наследственностью или свежей мутацией гена, отвечающего за метаболические процессы. Синдром де Тони-Дебре-Фанкони зачастую развивается не один, а совместно с цистинозом, галактоземией, синдромом Вильсона, первичной тирозинемией, непереносимостью фруктозы. Чаще эта форма фиксируется у детей, нежели у взрослых.
Провоцирующие факторы
 Острый дефицит витамина D может спровоцировать болезнь.
Острый дефицит витамина D может спровоцировать болезнь.
Синдром Фанкони с большей вероятностью появляется у людей со следующими наследственными нарушениями:
- сбой в процессах обмена цистина (аминокислоты в составе белков);
- непереносимость молочной продукции (даже грудного молока);
- дефекты различных ферментов, отвечающих за синтез и распад гликогена;
- сбой в обмене ароматических аминокислот;
- непереносимость фруктозы;
- проблемы с метаболизмом меди (болезнь Коновалова-Вильсона);
- нарушение обмена миелина и дисфункция фермента сульфатазы;
- постоянное воздействие токсинов на организм (лекарственных, ядов, тяжелых металлов);
- сбой в обменных процессах с участием белка, что приводит к отложению специфичного комплекса - амилоида;
- острый дефицит витамина D.
Врожденная и приобретенная
Синдром Фанкони - тяжелая рахитоподобная болезнь, которая может быть первичной (врожденной) и вторичной (приобретенной).
Врожденная патология де Тони-Дебре-Фанкони - наследственная, то есть передающаяся от родителей потомкам. Зачастую развивается совместно с другими генетическими нарушениями. Для состояния характерно острое нарушение метаболических процессов, в результате которых, наряду с дисфункцией почечных канальцев, развиваются слепота, синдром увеличенной печени, синдром со стойким снижением концентрации и тканевой активности гормонов щитовидки. Механизм наследования синдрома определяется типом патологии, совместно с которой он развивается.
Приобретенный синдром де Тони-Дебре-Фанкони зачастую провоцируется различными лекарствами:
- используемыми при лечении рака химиопрепаратами;
- антиретровирусными медикаментами;
- устаревшим тетрациклином.
Провокаторами развития вторичных форм патологии выступают осложненная трансплантация почки, онкологические заболевания крови, выраженные дефицитные состояния (преимущественно при нехватке витамина D). Если врожденное заболевание развивается еще внутриутробно и фиксируется в основном у детей, то вторичная форма может развиться и у взрослых.
Механизм развития и течения
 Нарушается обратное всасывание веществ в почках.
Нарушается обратное всасывание веществ в почках.
Синдром де Тони-Дебре-Фанкони вызван врожденной мутацией гена, отвечающего за метаболические процессы, но отмечаются случаи свежей трансформации, вызванной другими патологиями. Вследствие нарушения всасывания множества полезных веществ и ионов развиваются тяжелые дефицитные состояния:
- при нехватке аминокислот появляется выраженная дистрофия, замедляется рост и физическое развитие;
- при выведении фосфора и бикарбонатов - сбой процесса минерализации с началом разрушения костной ткани;
- при скоплении сахара в моче - нарушение регуляции углеводного обмена;
- при выведении с мочой калия - мышечная атрофия, падение АД до 80 мм РТ. ст. и ниже;
- при масштабных обменных нарушениях - деструкция почечной ткани (сужение просвета с уплощением эпителия и укорочением канальцев, в которых происходит обратное всасывание).
Симптомы у детей и взрослых
Первые проявления врожденного синдрома де Тони-Дебре-Фанкони появляются в первый год жизни, реже - с 1,5 лет. Малыш часто мочится, наблюдается субфибралитет (до 37-38°С), запор, рвота. К 6-ти годам при отсутствии лечения детки не ходят, а к 12-ти отказывают почки, что чревато смертью. Больные - необщительные, закомплексованные, пугливые. Умственная и мыслительная функция в норме. Последствия синдрома - проблемы с НС и зрением, развитие хронического иммунодефицита, дисфункция мочеполовых органов и ЖКТ.
Симптомы у детей развиваются по большей части в связи с дефицитом фосфатов (по типу фосфат-диабета) и проявляются:
- шаткостью походки;
- низким ростом;
- малой подвижностью;
- округленностью голеней;
- искривлением костей скелета (в частности, позвоночника);
- костными болями, отчего малыш не хочет ходить;
- резкое увеличение объемов выделяемой мочи;
- падение удельного веса на фоне качественного изменения состава мочи;
- мышечная атония;
- костные боли;
- гипертоническая болезнь;
- хроническая почечная недостаточность (при отсутствии лечения);
- остеопороз, остеопения на фоне падения плотности костной ткани.
Синдром де Тони - Дебре - Фанкони представляет собой тяжелое врожденное заболевание, характеризующееся разнообразными Страдают им чаще всего дети первого года жизни. Как правило, встречается в сочетании с другими наследственными патологиями, но может манифестировать и как самостоятельный синдром.
Краткий экскурс в историю
Заболевание было открыто и изучено в 1931 году доктором Фанкони из Швейцарии. Обследуя ребенка с рахитом, низким ростом и изменениями в анализах мочи, он пришел к выводу, что данное сочетание признаков стоит рассматривать как отдельную патологию. Спустя два года де Тони внес свои поправки, добавил к уже имеющемуся описанию гипофосфатемию, а еще через некоторое время Дебре выявил у подобных больных аминоацидурию.
В отечественной литературе это состояние называют терминами «наследственный синдром де Тони - Дебре - Фанкони» и «глюкоаминофосфатдиабет». За рубежом оно чаще именуется renal Fanconi syndrome.
Причины развития синдрома Фанкони
В настоящий момент так и не удалось до конца выяснить, что же лежит в основе этого тяжелого недуга. Синдром Фанкони предположительно является Специалисты считают, что развитие этой патологии связано с точечной мутацией, которая приводит к неправильной работе почек. Многочисленные исследования подтвердили, что в организме происходит нарушение клеточного обмена. Не исключено, что в деле замешан аденозинтрифосфат (АТФ) - соединение, играющее важную роль в обмене энергии. В результате некорректной работы ферментов теряется глюкоза, аминокислоты, фосфаты и другие не менее полезные вещества. В таких суровых условиях почечные канальцы не получают необходимой для их работы энергии. Полезные вещества выводятся вместе с мочой, нарушаются обменные процессы, развиваются рахитоподобные изменения костной ткани.
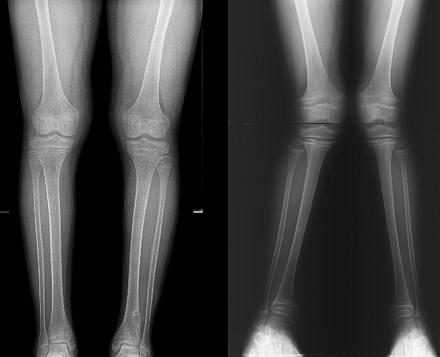
Синдром Фанкони у детей встречается гораздо чаще, чем у взрослых. По статистике частота патологии составляет 1:350 000 новорожденных. Болеют и мальчики, и девочки в равных пропорциях.
Признаки синдрома Фанкони
Заболевание может развиваться в любом возрасте, но чаще всего оно отмечается у детей первого года жизни. Глюкозурия, генерализованная гипераминоацидурия и гиперфосфатурия - эта триада признаков характеризует синдром Фанкони. Симптомы развиваются достаточно рано. Прежде всего родители замечают, что их ребенок начинает чаще мочиться, и его постоянно мучает жажда. Малыши, конечно же, не могут сказать об этом словами, но по их капризному поведению и постоянному зависанию на груди или бутылочке становится очевидно, что с ребенком что-то не так.

В дальнейшем родителям приносит много беспокойства частая беспричинная рвота, длительные запоры и не поддающаяся объяснению Как правило, на этом этапе ребенок наконец-то попадает на прием к врачу. Опытный педиатр может заподозрить, что на обычную простуду такое сочетание симптомов совсем не похоже. В случае если доктор окажется грамотным, он вовремя сможет распознать синдром Фанкони.
Симптомы между тем никуда не исчезают. К ним добавляется заметное отставание в физическом и умственном развитии, появляются выраженные искривления крупных костей. Обычно изменения затрагивают лишь нижние конечности, приводя к деформации по варусному или вальгусному типу. В первом случае ножки ребенка будут искривлены колесом, во втором - в виде буквы «Х». И тот и другой вариант, конечно же, неблагоприятны для дальнейшей жизни ребенка.
Синдром Фанкони у детей часто включает в себя остеопороз (преждевременное разрушение костной ткани), а также значительную задержку роста. Не исключены переломы длинных и параличи. Даже если до сих пор родители не беспокоились о состоянии малыша, то на этом этапе они уже точно не откажутся от квалифицированной помощи.

Синдром Фанкони у взрослых встречается довольно редко. Все дело в том, что это серьезное заболевание закономерно приводит к развитию почечной недостаточности. При таком раскладе невозможно дать какой-либо однозначный прогноз и гарантировать большую В литературе описаны случаи, когда в возрасте 7-8 лет синдром Фанкони сдавал свои позиции, наступало заметное улучшение состояния ребенка и даже выздоровление. К сожалению, такие варианты в современной практике достаточно редки, чтобы можно было сделать какие-то серьезные выводы.
Диагностика синдрома Фанкони
Кроме сбора анамнеза и тщательного осмотра доктор обязательно назначит некоторые обследования, позволяющие подтвердить данное заболевание. Синдром Фанкони неизбежно приводит к нарушению работы почек, а значит обязательным будет обычный анализ мочи. Конечно же, этого недостаточно, чтобы выявить все особенности течения болезни. Необходимо посмотреть не только содержание в моче белка и лейкоцитов, но и попытаться обнаружить лизоцим, иммуноглобулины и другие вещества. В анализе также обязательно выявится высокое содержание сахара (глюкозурия), фосфатов (фосфатурия), будут видны значительные потери важных для организма веществ. Подобное обследование можно провести как амбулаторно, так и в условиях стационара.

В анализах крови тоже неизбежны некоторые изменения. При биохимическом исследовании отмечается снижение почти всех значимых микроэлементов (прежде всего кальция и фосфора). Развивается выраженный мешающий нормальной работе всего организма.
Рентгенография скелета покажет остеопороз (разрушение костной ткани) и деформацию конечностей. В большинстве случаев обнаруживается отставание темпов роста костей и их несоответствие биологическому возрасту. При необходимости доктор может назначить УЗИ почек и других внутренних органов, а также обследование у смежных специалистов.
Дифференциальный диагноз
Бывают случаи, когда некоторые другие заболевания маскируются под синдром Фанкони. Перед врачом встает сложная задача разобраться, что же происходит с маленьким пациентом на самом деле. Иногда глюкоаминофосфатдиабет путают с хроническим пиелонефритом и другими заболеваниями почек. Изменения в анализах мочи, а также характерные особенности поражения костной ткани помогут педиатру выставить верный диагноз.
Лечение синдрома Фанкони
Стоит учесть тот факт, что данная патология является хронической. Довольно сложно полностью избавиться от неприятных симптомов, можно лишь на какое-то время уменьшить проявления болезни. Что же предлагает современная медицина в качестве помощи больным детям?
На первом месте стоит диета. Пациентам рекомендуют ограничить употребление соли, а также всех острых и копченых продуктов. В рацион добавляют молоко и различные фруктовые сладкие соки. Не стоит забывать и о (чернослив, курага и изюм). В том случае, когда дефицит микроэлементов дошел до критической стадии, врачи назначают прием специальных витаминных комплексов.
На фоне диеты вводят большие дозы витамина D. Состояние пациента постоянно контролируется - ему приходится время от времени сдавать кровь и мочу на анализы. Это необходимо для того, чтобы вовремя выявить начинающийся гипервитаминоз и снизить дозу витамина D. Лечение длительное, большими курсами, с перерывами. В большинстве случаев такая терапия помогает восстановить нарушенный обмен веществ и предотвратить тяжелые осложнения.

Если болезнь зашла далеко, пациент попадает в руки хирургов. Опытные ортопеды сумеют выправить костные деформации и существенно повысить уровень жизни ребенка. Подобные операции выполняются только в случае стойкой и длительной ремиссии: не менее полутора лет.
Прогноз
К сожалению, прогноз у таких пациентов неблагоприятный. В большинстве случаев болезнь медленно прогрессирует, рано или поздно приводя к почечной недостаточности. Деформации костей скелета неизбежно ведут к инвалидизации и ухудшению общего качества жизни.
Можно ли избежать этой патологии? Несомненно, подобный вопрос волнует каждого, кто столкнулся с синдромом Фанкони. Родители пытаются понять, что они сделали не так и где не уследили за ребенком. Не менее важно знать, грозит ли повторение ситуации с другими детьми. К сожалению, меры профилактики в настоящий момент не разработаны. Парам, планирующим обзавестись еще одним ребенком, стоит проконсультироваться у генетика для получения более полной информации о волнующей их проблеме.
Синдром Висслера - Фанкони (аллергический субсепсис)
Описано это заболевание только у детей от 4 до 12 лет. Причина этой серьезной патологии до сих пор не известна. Можно предположить, что этот синдром является типичным аутоиммунным заболеванием, особой формой ревматоидного артрита. Начинается всегда остро, с подъема температуры, которая неделями может держаться на цифре 39 градусов. Во всех случаях появляется полиморфная сыпь на конечностях, иногда на лице, груди или животе. Обычно происходит выздоровление без каких-либо серьезных осложнений. Однако же у части маленьких пациентов со временем развиваются тяжелые поражения суставов, приводящие к инвалидизации.
Ганкина Ю. В., ветеринарный врач, патолог, член ISFM. Ветеринарная клиника неврологии, травматологии и интенсивной терапии, г. Санкт-Петербург.Синдром Фанкони – наследуемая патология почек у собак породы басенджи, именно у этих пациентов большинство ветеринарных врачей проводят диагностику данного заболевания. Тем не менее описано множество случаев возникновения синдрома у собак других пород как первичной патологии, так и на фоне различных заболеваний или при приеме некоторых препаратов. Синдром Фанкони описан и у кошек.
Анатомия и физиология почек
Как известно, структурной и функциональной единицей почек является нефрон. Он начинается с шарообразной структуры – капсулы Боумена, состоящей из наружного и внутреннего листков. Наружный листок является продолжением эпителия почечного канальца. Внутренний слой сформирован высокоспециализированными клетками – подоцитами. Подоциты имеют множественные отростки-ножки, соприкасающиеся с ножками соседних клеток и покрывающие таким образом капилляры клубочка. Кровь в капилляры поступает через афферентную артериолу, которая делится на множество тонких петель, объединяющихся затем в выносящую эфферентную артериолу. Между эндотелием капилляров и подоцитами находится базальная мембрана, являющаяся важным звеном фильтрационного барьера.
Формирование мочи начинается с процесса ультрафильтрации. Весь объем плазмы фильтруется примерно 100 раз за день. Основными составляющими почечного фильтра являются эндотелий капилляров клубочков, базальная мембрана и подоциты внутреннего слоя капсулы Боумена. Большая часть гломерулярного фильтрата образуется за счет высокого гидростатического давления артериальной крови и выборочной проницаемости почечного барьера. Вода и растворенные в ней вещества проходят через фильтр, а крупные белковые молекулы удерживаются в кровяном русле. Таким образом получается очень большой объем почечного фильтрата. Далее в почечных канальцах происходит обратное всасывание необходимых компонентов. Около 99 % натрия хлорида и воды адсорбируется. Структура почечных канальцев коррелирует с их функцией. Так, например, эпителий проксимального отдела почечных канальцев имеет хорошо развитую щеточную каемку (для увеличения площади поверхности) и большое количество митохондрий (энергетический запас клеток). Основная часть транспортных функций, происходящих в данных клетках, является энергозависимой, поэтому они очень чувствительны к ишемическому воздействию. Кроме того, эпителий почечных канальцев может подвергаться токсическому воздействию экскретируемых веществ. В проксимальных канальцах активно реабсорбируется большое количество натрия и хлоридов, а вода всасывается пассивно и таким образом абсорбируется 60–80 % почечного фильтрата, поступающего в проксимальный почечный каналец. Канальцы и перитубулярные капилляры находятся в тесной близости для наиболее быстрого удаления абсорбированного натрия и воды. Кроме натрия и воды, в проксимальном отделе канальцев абсорбируются глюкоза, аминокислоты, фосфаты кальция, мочевая кислота, белки и калий.
Далее находится петля Генле, которая спускается из коркового вещества в мозговое и затем возвращается обратно. Толстая нисходящая часть петли Генле выстлана простым кубическим эпителием, тонкая нисходящая и восходящая части – простым плоским эпителием, микроворсинки у этих клеток выражены слабее или не выражены совсем, они содержат меньшее количество митохондрий. Тонкая часть петли Генле, тесно оплетенная капиллярами (прямые сосуды – vasa recta), играет основную роль в концентрации мочи. Далее следует толстая часть восходящей петли Генле, выстланная простым кубическим эпителием, напоминающим эпителий проксимальных канальцев, но с менее выраженными ворсинками, которые располагаются в области коркового вещества. Клетки, примыкающие к афферентной артериоле, являются составной частью юкстагломерулярного аппарата. После петли Генле располагается дистальный извитой каналец, он достаточно короткий и структурно напоминает эпителий проксимального извитого канальца, но с менее выраженными ворсинками, зато со значительным количеством митохондрий (иногда превышающих количество митохондрий в эпителии проксимального канальца). Следом за дистальным канальцем находится соединительный каналец, далее – собирательная трубочка. Собирательные трубочки являются продолжением нефрона, но имеют отличное происхождение. Основной функцией этого отдела мочевыделительной системы является концентрирование мочи за счет абсорбции воды под действием антидиуретического гормона.
Патофизиология
Синдромом Фанкони называют состояние, при котором развивается снижение реабсорбции в проксимальном отделе извитых канальцев почек. В результате этого большинство продуктов, которые должны возвращаться в кровоток после фильтрации первичной мочи, выводятся из организма. Происходит потеря с мочой глюкозы, фосфатов, аминокислот, бикарбоната, кальция, калия и других ионов. Нарушение всасывания бикарбоната приводит к состоянию, называемому почечным тубулярным ацидозом. Существует два типа почечного тубулярного ацидоза: проксимальный и дистальный.
При дистальном тубулярном ацидозе (тип 1) происходит нарушение закисления мочи. У таких пациентов часто развиваются нефролиты, происходит деминерализация костей, развивается гипокалиемия. Дефект проксимального отдела почечных канальцев приводит к нарушению реабсорбции бикарбонатов, такое состояние называют проксимальным тубулярным ацидозом (тип 2).
Пациенты с синдромом Фанкони также страдают потерей бикарбонатов, но, помимо этого, развивается потеря глюкозы, фосфатов, мочевой кислоты, аминокислот. Часто синдром Фанкони называют синдромом потери. При этом в моче отмечают повышенное содержание таких веществ, как глюкоза, натрий, калий, фосфор, бикарбонат и аминокислоты, в то время как в крови может отмечаться снижение этих показателей.
У животного с синдромом Фанкони часто обнаруживают глюкозурию при нормогликемии (если исключаются такие возможные причины, как лептоспироз и пиелонефрит), что является показанием для дальнейшей диагностики.
Существуют два различных типа синдрома Фанкони: врожденный и идиопатический. Врожденный синдром Фанкони встречается у собак породы басенджи.
Идиопатический синдром Фанкони описан у собак многих пород: бордертерьеров, норвежских элкхаундов, уиппетов, йоркширских терьеров, лабрадоров, грейхаундов, кокер-спаниелей некоторых окрасов, метисов (обычно имеются данные о единичных случаях).
Существует также приобретенный синдром Фанкони. Он развивается у собак при токсическом воздействии препаратов и различных веществ, поедании «китайских вкусняшек», при гипопаратиреозе, при болезнях печени, сопровождающихся накоплением меди.
У кошек описаны случаи возникновения приобретенного синдрома Фанкони при химиотерапии с применением хлорамбуцила.
Врожденный синдром Фанкони
Синдром Фанкони у басенджи – это наследственное заболевание, характеризующееся развитием недостаточности проксимального отдела канальцев почек. Впервые синдром Фанкони был описан в 1936 году швейцарским врачом Гвидо Фанкони. У собак первое описание синдрома Фанкони у басенджи датируется 1976 годом.
Синдром Фанкони басенджи вызван мутацией в гене FAN1 (Fanconi anemia-associated nuclease 1), который является частью семейства генов миотубуларин тирозинфосфатазы. Анемия Фанкони и синдром Фанкони вызваны мутацией в одном гене, но эти заболевания различны. При синдроме Фанкони мутация в гене FAN1 приводит к гиперчувствительности клеток проксимального отдела канальцев почек к минимальному количеству повреждающих веществ. Так как каждая индивидуальная собака может быть подвержена разному количеству различных повреждающих факторов, то и начало развития клинических симптомов может проявляться у животных разных возрастов. Обычно первые симптомы заболевания проявляются у животных в возрасте 4–7 лет. Патология всасывания натрия и фосфатов у басенджи развивается в чуть более раннем возрасте (3 лет), всасывание глюкозы и аминокислот снижается к 4 годам. Скорость клубочковой фильтрации может быть также нарушена.
Синдром Фанкони басенджи наследуется по аутосомно-рецессивному типу. От двух клинически здоровых родителей может быть получена больная собака, а оба родителя являются гетерозиготными. В этом случае 25 % потомства являются пораженными, 50 % – бессимптомными носителями и 25 % – здоровыми собаками без носительства мутантного гена. В настоящее время разработан тест для определения наличия мутации в гене FAN1. По различным данным, заболеваемость колеблется от 10 до 30 % среди собак породы басенджи.
Развитие синдрома Фанкони у собак с гепатопатией, сопровождающейся накоплением меди
Накопление меди может возникнуть вследствие как первичного нарушения метаболизма, так и нарушения экскреции меди на фоне заболеваний, связанных с холестазом. Выявление точной этиологии может быть проблематичным, так как при гистопатологическом исследовании будут обнаружены признаки фиброза, воспаления и цирроза вне зависимости от первоначальной причины.
Болезнь накопления меди (подобно болезни Вильсона у людей) описана у многих пород собак: вест-хайленд-уайт-терьер, доберман-пинчер, бедлингтонтерьер, скайтерьер, лабрадор-ретривер, далматин. Тем не менее генетическая основа обнаружена только у бедлингтонтерьеров, у которых накопление меди связано с дефектом в гене MURR-1.
Медь накапливается в печени, способствуя развитию гепатита, а в дальнейшем – цирроза печени, что еще больше усугубляет экскрецию меди. В процессе развития заболевания скопления меди отмечаются не только в паренхиме печени, но и в тканях почек и головном мозге. При поражении эпителия проксимального отдела почечных канальцев могут возникать признаки синдрома Фанкони. Данное состояние достаточно часто описывается у людей при развитии болезни Вильсона.
Для обнаружения меди в органах и тканях используют специальное окрашивание гистологических срезов. Поэтому для выявления данного состояния необходимо гистопатологическое исследование тканей печени и коркового вещества почек. Стоит отметить, что у людей с болезнью Вильсона не всегда удается обнаружить скопление меди в эпителии печени и почек. Это может быть связано с неравномерным распределением меди в паренхиме или диффузным наличием меди в цитоплазме, что не позволяет визуализировать ее при окрашивании. Также существуют методы количественной оценки меди в тканях.
Гипопаратиреоз
Описан случай развития синдрома Фанкони у собаки на фоне гипопаратиреоза. Точный механизм развития поражений проксимального отдела почечных канальцев неясен, но существует предположение, что он связан со снижением концентрации 1,25-дигидроксихолекальциферола в крови. У людей и крыс описано развитие синдрома Фанкони при недостаточности витамина D вне зависимости от уровня паратгормона. У данного животного снижение уровня 1,25-дигидроксихолекальциферола было вызвано гипофункцией паращитовидной железы. Симптомы недостаточности проксимального отдела почечных канальцев исчезли после заместительной терапии основного заболевания.
«Китайские вкусняшки»
В последние годы описано большое количество случаев возникновения приобретенного синдрома Фанкони после кормления собак лакомствами, произведенными в Китае. Чаще всего это собаки мелких пород, весом не более 10 кг. Случаи были зафиксированы в разных странах и на разных континентах: в Австралии, Северной Америке, Японии, Европе. Время от начала кормления данными продуктами до развития клинических симптомов различается от 0,3 до 78 недель. Предположительно это связано с количеством потребляемых лакомств, индивидуальной переносимостью, различиями в партиях и началом обнаружения владельцем клинических симптомов. Точный этиологический фактор еще не был определен. Предполагается, что им могут быть сальмонеллы, тяжелые металлы, пестициды, антибиотики, противовирусные препараты, микотоксины, родентициды, известные ранее нефротоксины или эндотоксины.
Отмечено развитие классических симптомов синдрома Фанкони, проходящих после отмены «китайских вкусняшек». Как правило, после симптоматического лечения и нормализации кормления животные полностью излечиваются, хотя и отмечались случаи развития хронической болезни печени.
Кошки
Синдром Фанкони крайне редко описывается у кошек. Отмечено развитие недостаточности проксимального отдела почечных канальцев у четырех кошек, проходящих лечение с применением хлорамбуцила. Хлорамбуцил был назначен в качестве химиотерапии при лечении алиментарной лимфомы или воспалительного заболевания кишечника. Симптомы синдрома Фанкони у данных пациентов развивались спустя 2–26 недель с начала химиотерапии. Немаловажно, что при этом ни у одной кошки не было симптомов полидипсии и полиурии, которые являются частыми проявлениями синдрома Фанкони у собак. Частичное или полное разрешение нефропатии отмечено в течение трех месяцев у 3 кошек из 4.
Симптомы синдрома Фанкони
Клинические проявления синдрома Фанкони могут сильно варьироваться по степени проявления и обычно включают в себя угнетение, рвоту, анорексию, обезвоживание, диарею, полидипсию, полиурию, потерю веса и плохое качество шерсти. Наиболее частый симптом, который отмечают владельцы, это полидипсия и полиурия (не описан у кошек).
Для подтверждения диагноза необходимо проведение биохимического анализа крови и мочи. При проведении лабораторных исследований наиболее распространенной находкой является глюкозурия при нормогликемии, что требует дальнейшего обследования. Следующим шагом для постановки диагноза является обнаружение аминоацидурии. Протеинурия обычно умеренная. Снижение реабсорбции бикарбоната приводит к развитию метаболического ацидоза. При длительном течении заболевания могут отмечаться гипокалиемия и мышечная слабость.
Лечение и прогнозы
Важным аспектом лечения синдрома Фанкони является исключение любых возможных причин. У пациентов с лептоспирозом, пиелонефритом могут пройти признаки недостаточности проксимального отдела канальцев почек в течение 2–3 месяцев после начала специфической терапии. Терапия синдрома Фанкони сводится к контролю метаболического ацидоза и замещению потерянных с мочой электролитов.
При развитии тяжелых изменений проводится госпитализация и агрессивная инфузионная терапия с целью нормализации состояния пациента.
У стабильных пациентов лечение может проводиться амбулаторно. Так как частыми симптомами являются полидипсия и полиурия, стоит уделять особое внимание нормальной гидратации пациента. У животных всегда должен быть доступ к свежей воде. При необходимости возможно подкожное введение жидкости стабильным пациентам, длительность терапии может составлять от нескольких недель до месяцев. Выбор раствора зависит от тяжести электролитных нарушений, как правило, необходимы подщелачивающие растворы, такие как раствор Рингера лактата, Нормосол-R, Плазмалит 156. В случае необходимости возмещения потерь калия проводится соответствующая инфузионная терапия в условиях отделения интенсивной терапии с постоянным контролем электролитов.
В случае развития тошноты и рвоты, отказа от еды необходимо применение антиэметиков, антацидных препаратов. У пациентов с уремией или кетонурией часто развиваются тошнота и отказ от корма. Также необходимо уделять внимание питанию. Не стоит снижать количество потребляемого белка у стабильных, неуремичных животных. Некоторые авторы рекомендуют дополнительные пищевые добавки, содержащие витамины и минеральные вещества, в качестве длительной нутриционной поддержки.
Развитие заболевания у басенджи может быть различным, по некоторым данным, синдром Фанкони может не влиять на продолжительность жизни больных собак. Прогрессия патологии у собак других пород, как правило, протекает достаточно быстро, но описаны и случаи спонтанного выздоровления. У одних собак хроническая почечная недостаточность может развиться в течение нескольких месяцев, другие остаются клинически стабильными в течение нескольких лет. Тем не менее чаще всего прогноз для животных с приобретенным синдромом Фанкони при проведении соответствующей терапии остается хорошим.
Список используемой литературы:Masaya Igase, Kenji Baba, Takako Shimokawa Miyama, Shunsuke Noguchi, Takuya Mizuno and Masaru Okuda. Acquired Fanconi syndrome in a dog exposed to jerky treats in Japan. J. Vet. Med. Sci. 77 (11): 1507–1510, 2015.
Natalie C Reinert and David G Feldman. Acquired Fanconi syndrome in four cats treated with chlorambucil. Journal of Feline Medicine and Surgery. 2016, Vol. 18(12) 1034–1040
Hill T. L., Breitschwerdt E. B., Cecere T. and Vaden S. Concurrent Hepatic Copper Toxicosis and Fanconi’s Syndrome in a Dog. J Vet Intern Med. 2008; 22: 219–222.
Ruth A. Darrigrand-Haag, Sharon A. Center, John F. Randolph, Robert M. Lewis and Philip A. Wood. Congenital Fanconi Syndrome Associated With Renal Dysplasia in 2 Border Terriers. Journal of Veterinary Infernal Medicine, Vol 70, No 6 (November-December), 1996: pp 412–419.
DiBartola S. Fluid, Electrolyte, and Acid-Base Disorders in Small Animal Practice, 4rd ed. St. Louis, MI: Saunders Elsevier, 2013.
Fanconi Renal Disease Management Protocol for Veterinarians By Steve Gonto, M. M. Sc., Ph. D. Revised February 29, 2016.
Ashley N. Hooper, DVM, Brian K. Roberts, DVM, DACVECC. Fanconi Syndrome in Four Non-Basenji Dogs Exposed to Chicken Jerky Treats. JAAHA | 47:6 Nov/Dec 2011.
Lisa M. Freeman, Edward B. Breitschwerdt, Bruce W. Keene, Bernie Hansen. Fanconi"s Syndrome in a Dog With Primary Hypoparathyroidism. Journal of Veterinary Internal Medicine, Vol8, No 5 (September-October), 1994: pp 349–354.
James C. M. Chan, Jon I. Scheinman, Karl S. Roth. Renal Tubular Acidosis. Pediatrics in Review Vol. 22 No. 8 August 2001, 277–287.
Appleman E. H., Cianciolo R., Mosenco A. S., Bounds M. E. and S. Al-Ghazlat. Transient Acquired Fanconi Syndrome Associated with Copper Storage Hepatopathy in 3 Dogs. J Vet Intern Med. 22: 1038–1042, 2008.
Roger A. Hostutler, DVM; Stephen P. DiBartola, DVM, DACVIM; Kathryn A. Eaton, DVM, PhD, DACVP. Transient proximal renal tubular acidosis and Fanconi syndrome in a dog. JAVMA, Vol 224, No. 10, May 15, 2004.
Hooijberg E. H., Furman E., Leidinger J., Brandstetter D., Hochleitner C., Sewell A. C., Leidinger E. and Giger U. Transient renal Fanconi syndrome in a Chihuahua exposed to Chinese chicken jerky treats. Tierarztl Prax Ausg K Kleintiere Heimtiere. 43(3): 188–192, 2015.
La Abraham, D Tyrrell Ja Charles. Transient renal tubulopathy in a racing Greyhound. Australian Veterinary Journal. Volume 84, No 11, November 2006.
Yearley J. H., et al. Survival time, lifespan, and quality of life in dogs with idiopathic Fanconi syndrome. J Am Vet Med Assoc. 225 (3): 377–383, 2004.










